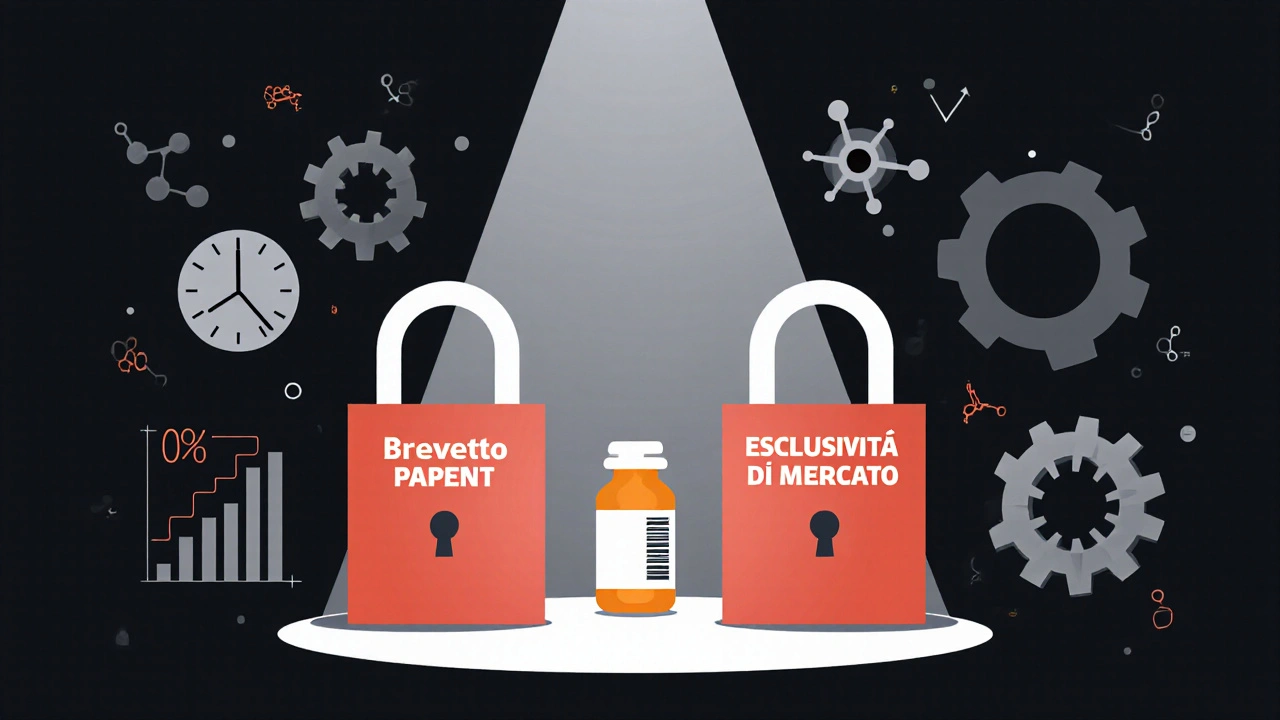
Se hai mai chiesto perché un farmaco costoso resta l’unico disponibile per anni, anche dopo che il brevetto è scaduto, la risposta sta in un dettaglio spesso trascurato: esclusività brevettuale e esclusività di mercato non sono la stessa cosa. Eppure, molte persone - anche nel settore farmaceutico - le confondono. E questa confusione ha un costo: miliardi di dollari in ritardi sull’ingresso dei generici, prezzi alti per i pazienti, e strategie aziendali che sfruttano un sistema più complesso di quanto sembri.
Cosa protegge davvero un farmaco?
Quando una azienda sviluppa un nuovo farmaco, ha due strumenti per proteggerlo dalla concorrenza: un brevetto e un periodo di esclusività di mercato. Ma mentre il brevetto è un diritto di proprietà intellettuale, l’esclusività di mercato è un privilegio regolatorio. Il primo lo rilascia l’Ufficio Brevetti (USPTO), il secondo lo concede la FDA. E non si sovrappongono come due cerchi, ma come due chiavi diverse che aprono la stessa porta.
Il brevetto protegge l’invenzione: la molecola, la sua formulazione, il modo in cui viene prodotta. Ha una durata standard di 20 anni dalla data di deposito. Ma qui c’è un inghippo: la maggior parte dei farmaci impiega 10-15 anni per arrivare sul mercato. Quindi, quando finalmente viene approvato, il brevetto potrebbe avere già 5-7 anni di vita. Il tempo effettivo di esclusiva sul mercato scende a 10-12 anni - spesso meno.
Per compensare, la legge permette di estendere il brevetto con la Patent Term Extension (PTE), ma con un tetto: non più di 5 anni di estensione, e mai oltre 14 anni dall’approvazione FDA. È un rimedio tecnico, non un diritto automatico. Devi richiederlo, dimostrare che il ritardo è stato causato dall’iter regolatorio, e non puoi estendere brevetti su prodotti già in commercio da anni.
L’esclusività di mercato: il potere nascosto della FDA
L’esclusività di mercato è un’altra cosa. Non ha nulla a che fare con l’invenzione. Non devi dimostrare che la molecola è nuova. Non devi nemmeno avere un brevetto. Basta che tu presenti dati clinici originali per ottenere un periodo in cui la FDA non può approvare farmaci generici o biosimilari, anche se sono chimicamente identici.
Questo è il cuore del sistema. Ecco i tipi principali:
- Esclusività per Nuova Entità Chimica (NCE): 5 anni. Durante questo periodo, la FDA non accetta nemmeno le domande dei generici. È il più potente: se hai sviluppato una molecola mai vista prima, hai 5 anni di protezione automatica, indipendentemente dai brevetti.
- Esclusività per farmaci orfani: 7 anni. Per malattie rare (meno di 200.000 pazienti negli USA). Anche se il farmaco non è brevettato, la FDA non approva concorrenti.
- Esclusività pediatrica: 6 mesi in più. Se l’azienda fa studi su bambini su richiesta della FDA, ottiene un’estensione di 6 mesi su qualsiasi protezione già in atto - brevetto o esclusività.
- Esclusività per biologici: 12 anni. Stabilita nel 2009 per i farmaci biologici (anticorpi, vaccini, terapie cellulari). È un periodo lungo, e non si può ridurre nemmeno se il brevetto scade prima.
- Esclusività di 180 giorni per il primo generico: Se un’azienda sfida un brevetto e vince, ottiene 180 giorni di esclusiva sul mercato dei generici. Un vantaggio enorme: può vendere il farmaco a prezzo più basso e guadagnare fino a 500 milioni di dollari in quel periodo.
Il caso del colchicina è emblematico. È un farmaco usato da secoli per la gotta. Nessun brevetto valido. Ma nel 2010, una società ha presentato nuovi dati clinici, ha ottenuto l’esclusività NCE, e ha fatto salire il prezzo da 10 centesimi a 5 dollari a compressa. Per 10 anni, nessun generico poteva entrare. Non perché fosse innovativo, ma perché la legge lo permette.
Perché la differenza conta davvero?
Perché un brevetto non si auto-applica. Se qualcuno produce un generico, devi andare in tribunale, pagare avvocati, aspettare anni. La FDA non blocca nulla di sua iniziativa. L’esclusività di mercato, invece, è un blocco automatico. La FDA rifiuta le domande dei concorrenti senza che tu debba fare nulla. È come un muro che si alza da solo.
Secondo i dati FDA del 2021, il 5,2% dei farmaci approvati aveva solo esclusività di mercato, senza alcun brevetto. E il 78% di questi non ha visto alcun generico durante l’intero periodo di protezione. Cioè: la FDA ha impedito la concorrenza, semplicemente perché la legge glielo ha imposto.
Allo stesso tempo, il 38,4% dei farmaci aveva solo brevetti. Ma se quei brevetti erano secondari - formulazioni, dosaggi, metodi d’uso - potevano essere facilmente aggirati. Un generico poteva usare la stessa molecola, ma con una formulazione diversa. E la FDA avrebbe approvato lo stesso. Senza esclusività, il brevetto da solo non basta.
Il sistema a doppia chiave
La legge Hatch-Waxman del 1984 ha creato un equilibrio: proteggere l’innovazione, ma non bloccare la concorrenza per sempre. E ha introdotto il concetto di “doppia chiave”: per entrare sul mercato, devi avere entrambe le chiavi. O almeno una delle due.
Quando un farmaco ha entrambi - brevetto ed esclusività - la protezione si sovrappone. Ma se uno scade, l’altro può continuare. E questo è il punto chiave. Molti pensano che quando il brevetto scade, il farmaco diventa generico. Non è vero. Se c’è ancora esclusività, il generico non può entrare. E molti generici aspettano anni, non perché il brevetto è in atto, ma perché l’esclusività della FDA è ancora valida.
Un esempio reale: Trintellix, un antidepressivo. Il brevetto principale è scaduto nel 2021. Ma la FDA aveva concesso 3 anni di esclusività NCE. Il primo generico è entrato solo nel 2024. Teva, che aveva preparato il prodotto da anni, ha perso 320 milioni di dollari in ricavi previsti.
Chi ne trae vantaggio?
Le grandi aziende lo sanno bene. Per loro, l’esclusività di mercato è spesso più preziosa del brevetto. Perché:
- Non richiede un’innovazione tecnica, solo dati clinici.
- Non può essere annullata da una causa legale come un brevetto.
- È automatica: non devi litigare, solo presentare la documentazione corretta.
Le piccole biotech, che spesso non hanno risorse per brevetti complessi, contano sull’esclusività NCE o orfana per proteggere i loro prodotti. Il 73% di loro si affida a questa protezione più che ai brevetti, secondo BIO nel 2023.
E poi c’è il lato economico: il 65% del fatturato di un farmaco viene generato nei primi 12 mesi dopo l’ingresso sul mercato, quando la protezione è al massimo. Per una molecola che costa 2,3 miliardi di dollari da sviluppare, quei 5-12 anni di esclusività non sono un lusso. Sono l’unica ragione per cui qualcuno investe.
Problemi e abusi
Il sistema funziona, ma è pieno di crepe. Le aziende usano i “brevetti secondari” per estendere il monopolio: brevetti su dosaggi, forme di somministrazione, o additivi. La FTC ha rilevato che il 68% dei brevetti elencati nell’Orange Book sono di questo tipo. Non proteggono l’invenzione, ma creano confusione.
E poi c’è il fenomeno dell’“evergreening”: quando un’azienda modifica leggermente un farmaco, presenta nuovi dati, e ottiene un’altra esclusività. È legale. Ma non è innovazione. È un modo per rinnovare il monopolio senza creare nulla di nuovo.
La FDA ha iniziato a reagire. Dal gennaio 2024, richiede giustificazioni più dettagliate per l’esclusività. E ha lanciato un nuovo strumento pubblico: l’Exclusivity Dashboard, che mostra in tempo reale quali farmaci sono protetti e per quanto tempo. È un passo verso la trasparenza. Ma non basta.
Quello che i farmacisti e i pazienti devono sapere
Se sei un paziente e ti chiedi perché un farmaco non ha generici, non guardare solo il brevetto. Guarda l’esclusività. Se il brevetto è scaduto, ma il farmaco è ancora costoso, potrebbe essere per via di un’altra protezione. Chiedi al tuo farmacista: “C’è ancora esclusività di mercato?”
Se sei un professionista del settore, non confondere i due concetti. Un brevetto scaduto non significa che il farmaco è libero. L’esclusività di mercato è un’altra bestia. E se non la gestisci bene, rischi di perdere anni di profitto - o di perdere milioni perché il generico arriva prima di quanto pensavi.
Il sistema è complesso, ma non è un mistero. È una macchina legale, costruita per bilanciare innovazione e accesso. Ma quando si perde di vista la differenza tra brevetto ed esclusività, la bilancia si spezza. E i pazienti pagano il prezzo.
Quale futuro per i farmaci?
Le tendenze sono chiare. Nel 2020, l’esclusività di mercato copriva il 41% del tempo totale di protezione dei farmaci. Nel 2027, secondo McKinsey, sarà il 52%. I brevetti stanno perdendo potere. I tribunali annullano sempre più brevetti secondari. Ma l’esclusività della FDA? Quella non si tocca. È più facile da difendere, più difficile da sfidare.
La proposta PREVAIL Act del 2023 vorrebbe ridurre l’esclusività per i biologici da 12 a 10 anni. È un segnale. Ma non cambierà il sistema alla radice. L’esclusività di mercato è diventata la nuova frontiera del monopolio farmaceutico.
La prossima volta che senti parlare di un farmaco “nuovo” o “rivoluzionario”, chiediti: è davvero innovativo? O è solo un vecchio principio attivo con una nuova etichetta, e un’altra esclusività di mercato che lo protegge?

Francesco Franceschelli
19.11.2025Io non sapevo che un farmaco potesse restare costoso anche dopo la scadenza del brevetto... questo mi fa venire i brividi. Non è giusto che la FDA tenga bloccati i generici per anni solo perché qualcuno ha presentato dati clinici su una molecola vecchia di 50 anni. E poi ci chiedono di fidarci del sistema.
Francesca Adduci
21.11.2025Lo sapevo. Da anni. Tutti lo sanno, ma nessuno parla. Le multinazionali comprano i politici, la FDA è un braccio operativo. Il colchicina? Un caso lampante. E tu pensavi che la sanità fosse per i pazienti.
Valentina Bonetto
21.11.2025Interessante il punto sull’esclusività pediatrica: 6 mesi in più su qualsiasi protezione esistente. È un incentivo intelligente, ma anche un’arma a doppio taglio. Se usata bene, migliora la salute dei bambini. Se usata male? Diventa un trucco per allungare i monopoli. Bisognerebbe definire meglio cosa conta come studio pediatrico utile.
Pietro Marinelli
22.11.2025MA CHE CAZZO!?!?!?!?!? E noi italiani paghiamo questi prezzi assurdi mentre negli USA fanno finta di regolamentare?!? Questo è un furto organizzato! La sanità non è un business, cazzo!! 😡
Anna Tarkia
23.11.2025È fondamentale distinguere tra data di deposito del brevetto e data di approvazione FDA: il gap di 10-15 anni è la norma, non l’eccezione. L’estensione del termine brevettuale (PTE) è un meccanismo di compensazione legittimo, non un abuso. L’esclusività di mercato, invece, è un incentivo regolatorio volto a stimolare la generazione di dati clinici indipendenti, non un privilegio arbitrario.
La FDA non blocca i generici per malizia, ma per rispetto del framework legislativo. L’Exclusivity Dashboard è un passo verso la trasparenza, non un’ammissione di colpa.
Marcello Fattoruso
23.11.2025Però è vero che il sistema funziona... solo che funziona troppo bene per chi ci guadagna. Io ho un parente che prende un farmaco per il diabete: 3 anni dopo la scadenza del brevetto, ancora niente generico. Il farmacista mi ha detto: "C'è ancora esclusività". E io pensavo fosse un errore. Invece no. È tutto legale. Ma non è giusto.
Martino Bonanomi
25.11.2025Quando leggo queste cose, mi chiedo: cosa significa davvero "innovazione"? Se un’azienda prende un farmaco di 70 anni, cambia la compressa da rotonda a ovale, aggiunge un colorante e ottiene 5 anni di esclusività... è innovazione o manipolazione? Forse il problema non è la legge, ma la nostra capacità di definire cosa sia un progresso reale. E non ci stiamo nemmeno provando.
Federico Righetto
26.11.2025Ma chi credete che paghi tutto questo? Noi! I contribuenti! La sanità pubblica italiana finanzia i farmaci cari, mentre le multinazionali americane fanno il botto! E voi vi limitate a commentare?!? Dobbiamo boicottare i farmaci USA, mandare lettere al ministro, fare manifestazioni! Non possiamo stare qui a guardare! 😤
Romeo india
27.11.2025Io ho fatto un’indagine su 12 farmaci. 9 avevano esclusività di mercato senza brevetto valido. 7 di questi erano vecchi farmaci rilanciati con un nuovo nome. Uno era un antinfiammatorio degli anni ’70. E adesso costa 12 euro a compressa. Non è un mercato. È un gioco da casino.
Danilo Domingos
28.11.2025Il fatto che il 78% dei farmaci con solo esclusività non abbia mai visto un generico... è sconcertante. Ma non è colpa della FDA. È colpa di noi che non chiediamo di più. Se i pazienti sapessero cosa cercare, se i farmacisti fossero più informati... forse si potrebbe cambiare qualcosa. Non serve una rivoluzione. Serve solo un po’ di consapevolezza.
Leonardo Chavez Medina
29.11.2025Forse il vero problema non è l’esclusività, ma il fatto che abbiamo trasformato la salute in un prodotto da mercato. Se un farmaco è vita, perché dobbiamo decidere chi può averlo in base a un brevetto? Non è un’idea di mercato che va cambiata, ma un’idea di umanità.
Matteo Bertozzi
1.12.2025Ho lavorato in un laboratorio di farmacologia per anni. Sapevamo che l’esclusività di mercato era il vero bottino. I brevetti? Erano la copertura. La vera battaglia era tra i team di ricerca che cercavano di produrre dati clinici sufficienti per ottenere l’NCE. Non per curare meglio. Per guadagnare di più. E il sistema lo permette. Non è malvagio. È semplicemente costruito così. E noi, come professionisti, abbiamo fatto finta di non vedere.