
Quando un farmaco generico arriva sullo scaffale di una farmacia, il paziente si aspetta che funzioni esattamente come il marchio originale. Ma cosa succede dietro le quinte? La risposta sta in un ambiente controllato con precisione: il cleanroom. Non è solo una stanza pulita. È un sistema complesso di filtri, pressioni, temperature e procedure umane che garantisce che ogni compressa, ogni iniezione, ogni goccia oculare sia priva di contaminanti che potrebbero uccidere o danneggiare chi la assume.
Cosa sono gli standard di cleanroom e perché contano per i farmaci generici
Gli standard di cleanroom non sono una scelta di lusso. Sono una richiesta legale. Negli Stati Uniti, la FDA li ha resi obbligatori con le norme cGMP (Current Good Manufacturing Practices) del 1978. In Europa, le regole sono scritte nell’Annex 1 dell’EudraLex, aggiornato nel 2023. Questi standard definiscono esattamente quanti particolati, batteri e virus possono essere presenti nell’aria di una stanza dove si producono farmaci. Per i farmaci generici, la sfida è ancora più grande. Non basta che il principio attivo sia lo stesso. Deve essere prodotto in modo che il corpo lo assorba esattamente come il farmaco originale. Se c’è una contaminazione, la sostanza potrebbe degradarsi. Se la polvere entra nel prodotto, potrebbe causare reazioni avverse. E se il prodotto non è sterile quando dovrebbe esserlo, può causare infezioni mortali. È successo nel 2012 con l’epidemia di meningite negli Stati Uniti, causata da un farmaco contaminato in un laboratorio non conforme. Da allora, i controlli sono diventati più severi.I quattro livelli di cleanroom: da A a D
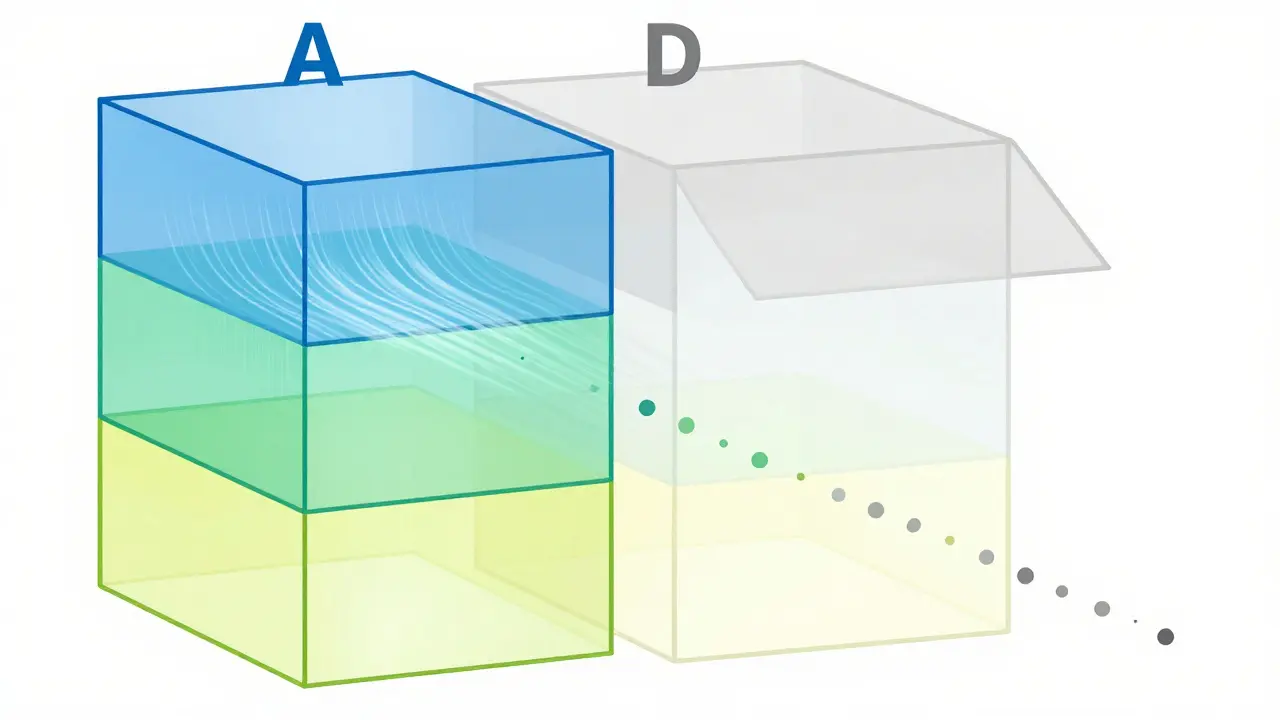
Non tutti i cleanroom sono uguali. Esistono quattro classi, o gradi, definite da ISO 14644-1, usate da tutto il mondo:- Grado A (ISO 5): È l’ambiente più pulito. Usato per le operazioni critiche come il riempimento di iniezioni sterili. Massimo 3.520 particelle per metro cubo di dimensioni ≥0,5 μm. L’aria fluisce in modo laminare, come un fiume senza turbolenze, a una velocità di 0,36-0,54 m/s. La pressione è più alta rispetto alle stanze vicine per evitare che l’aria sporca entri.
- Grado B (ISO 5 a riposo, ISO 7 in funzione): È l’area di supporto per il Grado A. Qui si preparano gli strumenti e si trasportano i materiali. Durante l’operazione, può contenere fino a 3,5 milioni di particelle. Richiede monitoraggio continuo.
- Grado C (ISO 7 a riposo, ISO 8 in funzione): Usato per operazioni meno critiche, come la preparazione di soluzioni o il confezionamento di compresse non sterili. Fino a 35 milioni di particelle in funzione.
- Grado D (ISO 8 a riposo): È il livello più basso. Usato per aree di stoccaggio o preparazione iniziale. Non ha limiti operativi, ma deve mantenere una pulizia minima.
Ogni grado ha anche limiti per i microrganismi. Nel Grado A, non devono esserci più di 1 unità formante colonie (CFU) per piatto di agar. Nel Grado D, si tollerano fino a 200 CFU. Questi valori non sono arbitrari. Sono basati su decenni di studi sulla sicurezza dei farmaci.
Le regole tecniche che fanno la differenza
Un cleanroom non è solo una stanza pulita. È un sistema vivente. Ecco cosa lo rende funzionale:- Filtri HEPA H13-H14: Catturano il 99,995% delle particelle da 0,3 μm. Sono come la retina di un occhio che non lascia passare neanche un granello di polvere.
- Numero di ricambi d’aria: Da 10 a oltre 60 volte all’ora, a seconda del grado. Nel Grado A, l’aria viene completamente sostituita ogni 20-30 secondi.
- Pressione differenziale: Le stanze più pulite hanno pressione più alta. L’aria esce, non entra. Se la porta si apre, l’aria scorre verso l’esterno, non verso l’interno.
- Temperatura e umidità: Devono essere tra 18-26°C e 30-60% di umidità. Troppo caldo o troppo secco fa evaporare i solventi. Troppo umido favorisce la crescita di muffe.
- Monitoraggio continuo: Sensori in tempo reale registrano particelle e batteri 24 ore su 24. Non basta fare un controllo una volta al giorno. Un piccolo sbalzo può significare un lotto da scartare.
Le differenze tra USA e Europa: un mondo di regole
La FDA non cita mai esplicitamente le classi ISO. Le sue norme dicono solo: “Progettate la struttura per evitare contaminazioni”. L’Unione Europea, invece, dice esattamente: “Grado A = ISO 5”. Questo crea un problema per i produttori che esportano. Un impianto che soddisfa la FDA potrebbe non essere accettato dall’EMA. Inoltre, gli Stati Uniti hanno norme diverse per le farmacie che preparano iniezioni (USP <797>). Lì, un’area tampone può essere ISO 7, mentre in un impianto di produzione di farmaci generici, lo stesso ambiente deve essere ISO 5. Questo significa che un produttore di generici deve spendere di più per lo stesso livello di sicurezza. Il Giappone richiede in più il monitoraggio delle particelle da 1,0 μm, un dettaglio che molti non considerano. Eppure, queste piccole differenze fanno la differenza quando la FDA ispeziona un impianto straniero.Il costo della pulizia: un peso per i produttori di generici
Costruire un cleanroom di Grado A costa tra $250 e $500 al metro quadrato. Per un impianto da 500 metri quadrati, si parla di oltre $125.000 solo per l’edificio. Poi ci sono i filtri da sostituire ogni 2-3 anni, i sensori da calibrare, il personale da addestrare. Il costo operativo annuo può superare il 20% del fatturato. I produttori di farmaci innovativi hanno margini del 70-80%. I produttori di generici? 15-20%. Questo significa che mentre i grandi laboratori possono permettersi di investire in tecnologie avanzate, le piccole aziende lottano per sopravvivere. Un produttore su Reddit ha raccontato di aver abbandonato la produzione di un’inalazione generica perché il costo di mantenere il Grado A rendeva impossibile guadagnare anche un centesimo su ogni unità. Ma il prezzo più alto non è quello in denaro. È il rischio di un richiamo. Nel 2022, Aurobindo Pharma ha dovuto ritirare $137 milioni di prodotti sterili perché il monitoraggio del Grado B non era adeguato. La FDA ha emesso 228 lettere di avvertimento quell’anno, il 63% per problemi di ambiente. Una sola violazione può chiudere un impianto per mesi.
Storie di successo e fallimento
Teva, uno dei più grandi produttori di generici, ha trasformato la sua linea di Copaxone. Ha installato sistemi isolatori nel Grado A, riducendo gli eventi di contaminazione da 12 a 2 all’anno. Prima, la FDA rifiutava la loro domanda. Dopo l’upgrade, l’approvazione è arrivata. Il costo? Milioni. Il ritorno? La fiducia dei pazienti e il mercato globale. Al contrario, un piccolo produttore indiano ha speso $4,2 milioni per aggiornare il suo impianto agli standard dell’Annex 1. Ma i suoi sistemi di ventilazione non erano adatti al clima tropicale. L’umidità ha causato condensa nei filtri. I sensori hanno segnalato errori. La FDA ha bloccato le esportazioni. Il costo dell’aggiornamento è diventato un peso insostenibile.Cosa cambierà nei prossimi anni
L’Annex 1 del 2023 ha introdotto nuove regole: monitoraggio continuo obbligatorio, campionamento dell’aria più frequente, strategie di controllo della contaminazione documentate in modo dettagliato. La FDA ha detto che seguirà. Il prossimo anno, pubblicherà linee guida allineate. Le nuove tecnologie stanno arrivando. Robot che indossano tute e trasportano materiali senza contatto umano. Intelligenza artificiale che prevede quando un filtro si intasa. Sistemi a uso singolo che riducono la necessità di pulizia. McKinsey stima che entro il 2028, l’automazione ridurrà i costi operativi del 25-30%. Ma il vero cambiamento sarà nei farmaci stessi. I biosimilari, le terapie avanzate, gli inalatori complessi richiedono ambienti più controllati. La FDA prevede che nel 2025, il 50% delle domande di farmaci generici richiederà Grado A o B. Non è più un’opzione. È la nuova normalità.Come prepararsi: consigli pratici per i produttori
Se sei un produttore di farmaci generici, non puoi aspettare che la FDA ti chiami. Ecco cosa devi fare:- Classifica il tuo processo: Quale grado ti serve? Un’inalazione? Un’iniezione? Una compressa orale? Ogni prodotto ha un livello richiesto.
- Investi nel monitoraggio: Non basta un controllo manuale ogni settimana. Servono sensori in tempo reale con allarmi automatici.
- Addestra il personale: Il 42% degli errori viene dai protocolli di vestizione. Un operatore che tocca la maschera con le mani guantate può contaminare un intero lotto.
- Documenta tutto: La FDA non chiede solo risultati. Chiede prove. 15-20 procedure operative standard, con firme, date, e registrazioni di calibrazione.
- Usa le risorse gratuite: La FDA ha moduli di formazione online. ISPE e PDA pubblicano guide tecniche. Non devi partire da zero.
La qualità di un farmaco generico non si misura solo nella sua composizione chimica. Si misura nell’aria che lo circonda. Nella pressione della stanza. Nella disciplina di chi lo tocca. Nella precisione di un sensore che non si è fermato per un solo secondo. Perché quando un paziente prende una pillola, non sa se è generica o di marca. Ma sa che deve funzionare. E che non deve ucciderlo.
